
Markery DNA związane z miejscem ograniczenia - Restriction site associated DNA markers
Markery DNA związane z miejscami restrykcyjnymi (RAD) są rodzajem markerów genetycznych, które są przydatne do mapowania asocjacyjnego, mapowania QTL , genetyki populacyjnej , genetyki ekologicznej i genetyki ewolucyjnej. Wykorzystanie markerów RAD do mapowania genetycznego jest często nazywane mapowaniem RAD. Ważnym aspektem markerów RAD i mapowania jest proces izolowania znaczników RAD, które są sekwencjami DNA, które bezpośrednio flankują każde wystąpienie określonego miejsca restrykcyjnego enzymu restrykcyjnego w całym genomie. Po wyizolowaniu znaczników RAD można je wykorzystać do identyfikacji i genotypowania polimorfizmów sekwencji DNA, głównie w postaci polimorfizmów pojedynczego nukleotydu (SNP) . Polimorfizmy, które są identyfikowane i genotypowane przez izolację i analizę znaczników RAD, określane są jako markery RAD.
Izolacja tagów RAD
Zastosowanie flankujących sekwencji DNA wokół każdego miejsca restrykcyjnego jest ważnym aspektem znaczników RAD. Gęstość znaczników RAD w genomie zależy od enzymu restrykcyjnego użytego podczas procesu izolacji. Istnieją inne techniki markerów miejsc restrykcyjnych, takie jak RFLP lub polimorfizm długości amplifikowanych fragmentów (AFLP), które wykorzystują polimorfizm długości fragmentów spowodowany różnymi miejscami restrykcyjnymi do rozróżniania polimorfizmu genetycznego. Zastosowanie flankujących sekwencji DNA w technikach znaczników RAD jest określane jako metoda zredukowanej reprezentacji.
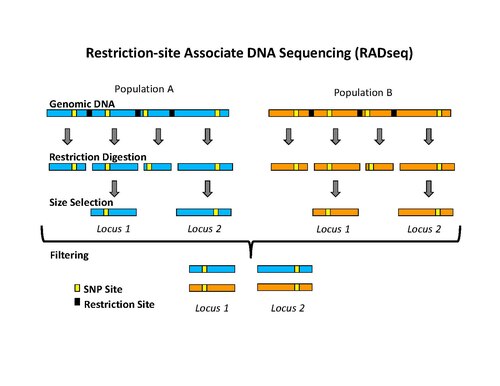
Początkowa procedura izolowania znaczników RAD obejmowała trawienie DNA określonym enzymem restrykcyjnym, ligację biotynylowanych adaptorów z nawisami, losowe cięcie DNA na fragmenty znacznie mniejsze niż średnia odległość między miejscami restrykcyjnymi oraz izolowanie biotynylowanych fragmentów za pomocą kulek streptawidyny . Procedurę tę zastosowano początkowo do wyizolowania znaczników RAD do analizy mikromacierzy . Niedawno procedura izolacji znaczników RAD została zmodyfikowana do użytku z wysokoprzepustowym sekwencjonowaniem na platformie Illumina , co ma tę zaletę, że znacznie zmniejszono surowe wskaźniki błędów i wysoką przepustowość. Nowa procedura polega na trawieniu DNA określonym enzymem restrykcyjnym (na przykład: SbfI, NsiI,…), ligacji pierwszego adaptera, zwanego P1, do nawisów, losowym dzieleniu DNA na fragmenty znacznie mniejsze niż średnia odległość między miejscami restrykcyjnymi, przygotowanie ściętych końców na tępe końce i ligację drugiego adaptera (P2) oraz zastosowanie PCR do specyficznej amplifikacji fragmentów zawierających oba adaptery. Co ważne, pierwszy adapter zawiera krótki kod kreskowy sekwencji DNA, zwany MID (identyfikator molekularny), który jest używany jako marker do identyfikacji różnych próbek DNA, które są łączone i sekwencjonowane w tej samej reakcji. Zastosowanie wysokoprzepustowego sekwencjonowania do analizy znaczników RAD można zaklasyfikować do sekwencjonowania o zmniejszonej reprezentacji, do którego zalicza się m.in. RADSeq (RAD-Sequencing).
Wykrywanie i genotypowanie markerów RAD
Po wyizolowaniu znaczników RAD można je wykorzystać do identyfikacji i genotypowania polimorfizmów sekwencji DNA, takich jak polimorfizmy pojedynczego nukleotydu (SNP). Te polimorficzne miejsca są określane jako markery RAD. Najskuteczniejszym sposobem znalezienia znaczników RAD jest wysokoprzepustowe sekwencjonowanie DNA , zwane sekwencjonowaniem znaczników RAD, sekwencjonowaniem RAD, RAD-Seq lub RADSeq.
Przed opracowaniem wysokowydajnych technologii sekwencjonowania markery RAD były identyfikowane poprzez hybrydyzację znaczników RAD z mikromacierzami. Ze względu na niską czułość mikromacierzy, to podejście może wykryć jedynie polimorfizmy sekwencji DNA, które zakłócają miejsca restrykcyjne i prowadzą do braku znaczników RAD lub znaczne polimorfizmy sekwencji DNA, które zakłócają hybrydyzację znaczników RAD. Dlatego gęstość markerów genetycznych, którą można osiągnąć za pomocą mikromacierzy, jest znacznie niższa niż w przypadku wysokoprzepustowego sekwencjonowania DNA.
Historia
Markery RAD zostały najpierw zaimplementowane przy użyciu mikromacierzy, a później zaadaptowane do NGS (Next-Generation-Sequencing). Został on opracowany wspólnie przez laboratoria Erica Johnsona i Williama Cresko na Uniwersytecie w Oregonie około 2006 roku. Potwierdzili przydatność markerów RAD, identyfikując punkty przerwania rekombinacji u D. melanogaster i wykrywając QTL u cierników.
ddRADseq
W 2012 roku zaproponowano zmodyfikowaną metodę znakowania RAD o nazwie double digest RADseq (ddRADseq). Poprzez dodanie drugiego enzymu restrykcyjnego, zastąpienie losowego ścinania i ścisły etap selekcji rozmiaru DNA, możliwe jest przeprowadzenie taniego genotypowania populacji. Może to być szczególnie potężne narzędzie do skanowania całego genomu w celu selekcji i różnicowania populacji lub adaptacji populacji.